파이썬으로 이거 가능한지 여쭤보고 싶습니다.
글쓴이: riodiv / 작성시간: 토, 2016/08/27 - 1:17오후
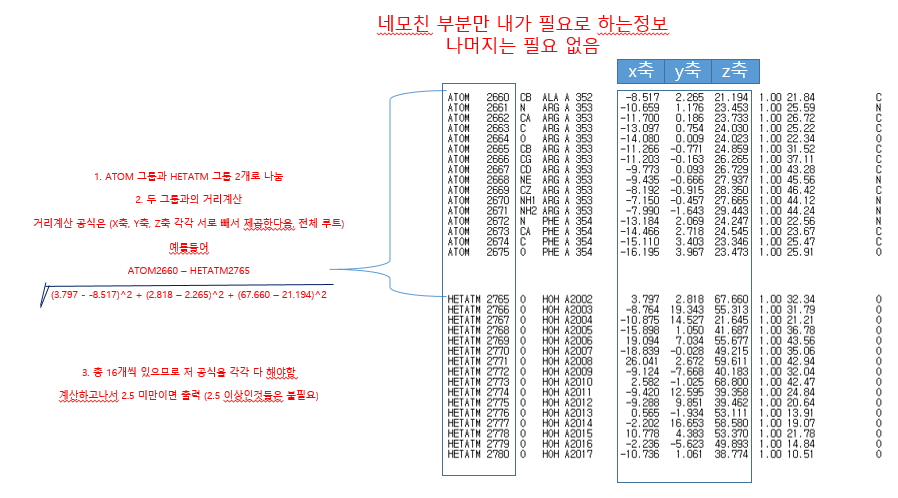
안녕하세요. 생물정보학을 전공하고 있습니다. (아마 생물쪽 분들은 안계시니깐 그냥 파일 작업만 여쭤볼려고 합니다)
파일 첨부했는데요. 보시다시피 파일자체는 저렇게 질서 정렬하게 되어 있습니다.
파이썬으로 저 작업 가능한지 알고 싶습니다. (다른언어가 아닌 파이썬으로 해야합니다 ㅠ)
file=open
이거해서 파일 연다음에.. 그담에 어떻게 해야할지 모르겠네요 ㅠㅠ
File attachments:
| 첨부 | 파일 크기 |
|---|---|
| 2.72 KB | |
| 257.45 KB |
{kind=link}
Forums:
당연히 가능은 합니다만 질문을 하신 것으로 보아,
당연히 가능은 합니다만 질문을 하신 것으로 보아, 두가지 문제가 있네요.
1. 기본적으로 python으로 할 수 있느냐 보다, logic 을 어떻게 만들어야 할지 자체를 모르시는 것 같고요.
2. raw data에서 ATOM과 HETATM 간의 연계를 어떻게 할 것인가 (즉 두 데이터간의 연결 정보가 애매모호 하다는 점)
이 두가지에 따라 가능하냐 못하냐의 문제인 듯 싶습니다.
일단, python으로 할 수 있느냐 없느냐만 말하자면, raw data의 문제가 없다면 당연히 가능 합니다.
기본적으로는 파일을 열고, 한 라인씩 읽어 들여 split을 이용하여 각 filed를 배열로 만든 다음, ATOM과 HETATM을 각각의 배열로 저장을 합니다.
ATOM, HETATM 배열이 다 만들어 지면 배열에 있는 정보로 연산을 하시면 되는데, 문제는 ATOM과 HETATM을 연산할 키가 없어 보이기는 합니다. 순서대로 하면 된다면, 배열 순서를 키로 잡아서 연산을 하면 되는데, 그것 까지는 알 수가 없네요.
단순 로직을 짜 보자면
file open loop util file end -> read per 1 line -> split field (split_result) -> if (split_result[0] == ATOM) atom[no] = split_result else hetatm[no] = split_result loop end loop 0 to until array_number-1 -> calculation atom[no] - hetatm[no] ~~ (루트 연산은 sqrt method를 사용하시면 될 것 같고..)이런 식으로 만드시면 됩니다. 위의 경우는 atom과 hetatm의 공통 키가 없어서, 라인 순서대로라고 가정을 한 로직 입니다.
감사합니다^^
로직을 설명 안드린 이유는, 저게 생물, 화학 분야라서 그렇습니다.
원래 베이스가 컴퓨터 전공한분들이 설명해주면 더 이해하기 힘드실까봐...
Atom 은 단백질을 구성하는 원자이고, Hetatom (헤테로 원자)는 물분자를 구성하는 원자입니다.
그래서 단백질을 구성하는 모든 원자와 물분자들의 모든 거리를 구하고자 하는것이 목적이었습니다.
거리가 너무 먼 것(2.5 이상)은 필요없어서 제외시키는 것입니다.
그 logic을 말한 것이 아니라 프로그밍을 어떻게
그 logic을 말한 것이 아니라 프로그밍을 어떻게 할 것인지에 대한 logic을 말씀 드린 것입니다. 다른 언어로 님의 문제를 해결할 수 있다면 제가 상황을 잘못 판단한 것이고요.
그리고, "거리가 너무 먼 것(2.5 이상)은 필요없어서 제외시키는 것"라는 규칙이 raw data에서 구현이 가능 하다면 프로그래밍적으로 가능할 것이고, 이 규칙을 raw data를 이용하여 만들 수 없다면 python이 아니라 어떤 언어로도 구현이 불가능 할 것 같다는 것 입니다.
혹시 SciPy 같은걸 찾고 계신게 아닌가 싶은데요
혹시 SciPy 같은걸 찾고 계신게 아닌가 싶은데요
Scientific Computing Tools for Python
https://www.scipy.org/about.html
-- Signature --
青い空大好き。
蒼井ソラもっと好き。
파란 하늘 너무 좋아.
아오이 소라 더좋아.
#!/usr/bin/env python3
해당 색인과 결과값이 일치하는지 출력된 결과행에서
소스코드 원본: https://gitlab.com/soyeomul/test/raw/master/155976.py
해당 색인과 결과값이 일치하는지 출력된 결과행에서 2개를 찝어서,
실제 계산기로 두들겨서 검산을 해봤씁니다^^^
인덱스를 추출하는 산법(알고리즘)이 좀 무식했는데...
좀 더 세련된 방법을 배우고 싶습니다.
꾸벅,,,
황병희 드림
[ibus-hangul(서라운딩 패치판)에서 작성했씁니다]
--
^고맙습니다 감사합니다_^))//
댓글 달기